Синдром барттера что это
Врожденные нарушения транспорта в почечных канальцах формируют спектр редких состояний, при каждом из которых поражаются определенные сегменты нефрона. Успехи генетики и молекулярной биологии позволили расшифровать патогенез многих таких заболеваний и углубили наши представления о регуляции водно-электролитного обмена в норме.
Синдром Бартера редкая форма гипокалиемического метаболического алкалоза с гиперкальциурией наследуется аутосомно-рецессивно. Различают два клинических подтипа синдрома Бартера. Антенатальный синдром Бартера (называемый также синдромом гиперпродукции простагландина Е) обычно проявляется у новорожденных и протекает тяжелее, чем классический синдром Бартера; он включает многоводие в анамнезе, потерю соли и выраженное обезвоживание.
Более легкий классический фенотип проявляется позднее задержкой развития ребенка и частыми эпизодами обезвоживания в анамнезе. Фенотипически сходный синдром Гительмана обусловлен другим генетическим дефектом. Описан также вариант антенатального синдрома Бартера с нейросенсорной глухотой и ХПН, имеющий другую генетическую основу.
Патогенез синдрома Бартера у детей. Биохимические сдвиги при синдроме Бартера (гипокалиемический метаболический алкалоз с гиперкальциурией) напоминают последствия применения петлевых диуретиков и отражают нарушение транспорта натрия, хлорида и калия в восходящем отделе петли Генле. Потеря натрия и хлорида, приводящая к уменьшению внутрисосудистого объема, стимулирует ренин-ангиотензин-альдостероновую систему.
Альдостерон усиливает реабсорбцию натрия и секрецию калия, тем самым усугубляя гипокалиемию. Он усиливает также секрецию ионов водорода в дистальных отделах нефрона, что усугубляет метаболический алкалоз. Гипокалиемия стимулирует синтез простагландинов, которые еще больше активируют ренин-ангиотензин-альдостероновую систему. В основе синдрома Бартера лежат три разных генетических дефекта транспортеров, функционирующих на уровне петли Генле.
Каждый из них тем или иным образом участвует в транспорте натрия и хлорида. При антенатальном синдроме Бартера обнаруживаются мутации гена, кодирующего натрий/калий/2 хлоридный транспортер NKCC2 (объект действия фуросемида), или гена люминальных калиевых каналов (ROMK), тогда как для классического синдрома характерны дефекты базолатеральных хлоридных каналов (ClC-Kb).
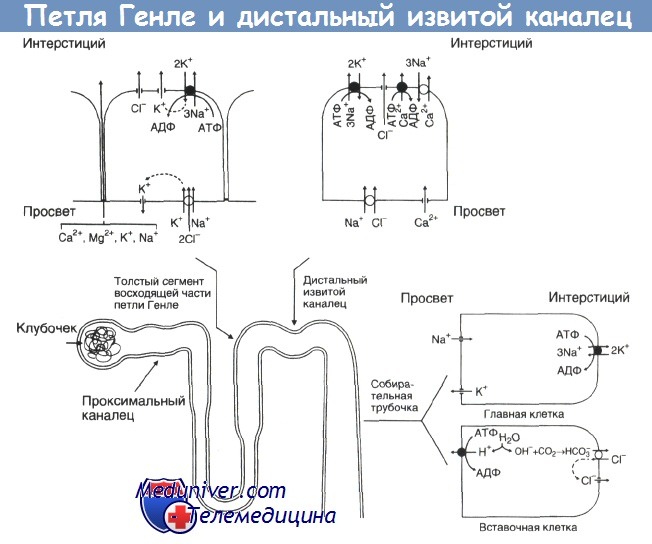 Основные переносчики апикальной и базолатеральной мембраны толстого сегмента восходящей части петли Генле, дистального извитого канальца и коркового отдела собирательной трубочки.
Основные переносчики апикальной и базолатеральной мембраны толстого сегмента восходящей части петли Генле, дистального извитого канальца и коркового отдела собирательной трубочки.
Клинические проявления синдрома Бартера у детей. Как выясняется из анамнеза, при беременности было многоводие. У новорожденного возможна дизморфия, например треугольное лицо, оттопыренные уши, косоглазие и опущение углов рта. Родственная связь между родителями указывает на аутосомно-рецессивное наследование синдрома. Для более позднего возраста характерны повторные эпизоды обезвоживания, задержка развития и биохимические нарушения. При синдроме Бартера всегда имеют место гипокалиемия и метаболический алкалоз.
Содержание кальция в моче обычно повышено. Часто значительно повышен уровень ренина, альдостерона и простагландина Е в сыворотке крови, особенно при тяжелой антенатальной форме синдрома. АД в большинстве случаев нормальное, хотя обезвоживание при выраженной потере соли у больных с антенатальной формой синдрома может приводить к артериальной гипотонии. Функция почек, как правило, сохранена. При УЗИ иногда обнаруживается нефрокальциноз как следствие гиперкальциурии.
Диагностика синдрома Бартера у детей. Диагноз устанавливают на основании клинической картины и лабораторных данных. У новорожденных о синдроме Бартера свидетельствует гипокалиемия (обычно ниже 2,5 ммоль/л) на фоне метаболического алкалоза. В типичных случаях отмечается гиперкальциурия. Гипомагниемию обнаруживают лишь у немногих больных; она более характерна для синдрома Гительмана. Поскольку проявления напоминают последствия продолжительного использования петлевых диуретиков, всегда необходимо выяснить, применялись ли эти средства (даже у маленьких детей).
Аналогичная клиническая картина имеет место при хронической рвоте, но при этом содержание хлорида в моче снижено, тогда как при синдроме Бартера оно повышено. При гистологическом исследовании почек находят гиперплазию юкстагломерулярного аппарата. Однако диагностическая биопсия при синдроме Бартера выполняется редко.
Лечение и прогноз синдрома Бартера у детей. Терапия синдрома Бартнера направлена на предотвращение обезвоживания и поддержание питания и коррекцию гипокалиемии. Зачастую требуются очень большие дозы калия, но и в этих случаях его уровень в сыворотке не всегда удается нормализовать, особенно у новорожденных. Грудные и маленькие дети могут нуждаться и в добавках натрия. Эффективен также индометацин, ингибирующий синтез простагландинов.
При внимательном отношении к электролитному балансу, объемному статусу и росту ребенка долговременный прогноз обычно благоприятный. Однако хроническая гипокалиемия, нефрокальциноз, длительное введение индометацина могут иногда приводить к развитию интерстициального нефрита и ХПН.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Синдром Барттера
Синдром Барттера — заболевание, которое передается на генетическом уровне и проявляется в виде гипокалиемии (нарушение электролитных обменных процессов), гиповолемии, метаболического алкалоза (сбой кислотно-щелочного равновесия), вторичного гиперальдостеронизма и компенсаторной гиперплазии юкстагломерулярного отдела почек.
Синдором Барттера проявляется с самого рождения, а при ухудшении состояния наблюдается развитие нефрокальциноза, что в итоге приводит к почечной недостаточности. Обнаружить данное заболевание можно по следующим признакам:
В ходе урологических исследований специалисты заметили, что синдром Барттера является генной мутацией дефекта петли Генле. Это в своем роде наследственность согласно аутосомно-рецессивному типу. Почечные нефроны при синдроме Баттера не способны удерживать калий, поэтому он быстро выводится из организма вместе с мочой, а циркуляция крови при этом значительно уменьшается в объеме как при пониженном, так и при нормальном АД.
Синдром Барттера классифицируют по видам согласно пораженным генам, в зависимости от характеристик их делят на:
Причины синдрома Барттера
Главная причина возникновения синдрома Барттера заключается в неправильности функционирования почечных канальцев, а именно процессов транспортизации. Такое нарушение проявляется через сокращение клетками восходящей части петли Генле реасорбции ионов Cl и Na, это поводит к:
Встречается также синдором псевдо-Барттера, но он никак не связан с генетическими мутациями, а вызван муковисцидозом, продолжительным диетическим питанием, длительным приемом слабительных препаратов и диуретиков.
Следственные связи причин синдрома Барттера и их особенности:
Симптомы синдрома Барттера
Симптоматика синдрома Баттера обнаруживается специалистами сразу после рождения или в первые года жизни ребенка. Заболевание характеризуется хронической нехваткой калия в организме, так как он быстро выводится. Основные симптомы следующие:
Касательно типов синдрома Барттера, то неонатальный возникает еще в период внутриутробного развития при многоводии. Беременность женщины, у которой плод имеет данное заболевание, протекает достаточно сложно и чаще всего оканчивается преждевременными родами. В большинстве случаев у детей врачи отмечают сонливость, отсутствие аппетита, плохой набор веса или его потерю, мышечную гипотонию, гипертермию, нарушение психомоторного развития, слуха и зрения.
Классический тип синдрома Барттера начинает проявляться после 1-го года жизни в виде:
Последний 6-й тип (синдром Гительмана) синдрома Барттера становится явным после шестилетнего возраста, при этом у ребенка наблюдается сильная утомляемость, мышечная слабость, однако, течение заболевания отличается большей мягкостью, нежели у других типов.
Диагностика синдрома Барттера
Диагностируют синдром Барттера согласно присутствию мышечной гипотонии и полиурии в клинической картине пациента еще в детском возрасте. Проявление заболевания заключается в следующем:
Лабораторные анализы и проведение врачебной диагностики организма дают возможность убедиться в наличии или отсутствии у пациента синдрома Барттера, в редких случаях проводят биопсию почек. Она позволит проявиться гиперплазии в области околоклубочкового аппарата. Не стоит путать синдром Барттера с последствиями, связанными с частым употреблением препаратов мочегонного действия, хронической рвотой, дефицитом магния, надпочечниковой недостаточностью и гиперальдостеронизмом изолированного типа.
Лечение синдрома Барттера
При лечении синдрома Барттера применяют как медикаментозную, так и заместительную терапию. Главной задачей врачей будет обеспечение пациента необходимым количеством хлорида натрия и калия. Важно соблюдать диету, обогащенную вышеуказанными веществами и специальными препаратами.
К заместительной интенсивной терапии неонатального типа синдрома Барттера приступают мгновенно после рождения, при этом врачи используют инфузии солевых растворов и калиесберегающие диуретики. Важно включить ингибиторы синтеза простагландинов и АПФ, ведь это позволит понизить секрецию альдостерона и ренина. При недоношенности новорожденного применение индометацина нужно отсрочить на 4-6 недель, так как появления сложных побочных эффектов не избежать. Для коррекционного лечения синдрома Гительмана используют препараты магния, а при синдроме псевдо-Барттера специалисты выявляют основные причины и устраняют их.
Прогноз синдрома Барттера
Если клиническое течение отличается длительностью и тяжелым состояние пациента, тогда синдром Барттера начинает сопровождаться нефрокальцинозом, который в большинстве случаев приводит к хронической недостаточности почек.
Синдром Барттера

Синдром Барттера – это генетически обусловленная тубулопатия, проявляющаяся выраженными нарушениями электролитного обмена (гипокалиемией), кислотно-щелочного равновесия (метаболическим алкалозом), гиповолемией, компенсаторной гиперплазией юкстагломерулярного (околоклубочкового) аппарата почек и вторичным гиперальдостеронизмом. Диагностируется по клинической симптоматике: полиурии, отставании в психомоторном развитии, гипотонии мышц, а также лабораторным показателям крови и мочи. Лечение заключается в заместительной терапии препаратами калия, натрия и магния, приеме калийсберегающих диуретиков, ингибиторов синтеза простагландинов и АПФ.

Общие сведения
Причины
Причиной синдрома Барттера считают нарушение транспортной функции почечных канальцев, проявляющееся снижением реабсорбции ионов Cl (и, соответственно, Na) клетками восходящего отдела петли Генле. Это приводит к гиповолемии, избытку натрия и воды в дистальной части нефрона, усилению секреции ионов K и натрий-калиевого обмена. Гипокалиемия стимулирует, в свою очередь, образование простагландинов Е2 и I2, приводящее к усилению секреции ренина и ангиотензина II.
Хроническая гиперренинемия способствует развитию гиперплазии юкстагломерулярного аппарата почек и повышенной продукции альдостерона надпочечниками. Ангиотензин II и альдостерон вызывают увеличение уровня почечного калликреина с дальнейшим повышением содержания брадикинина плазмы крови. Альдостерон приводит к усилению выведения калия почками. Калликреин (брадикинин) и простагландины блокируют вазопрессорный эффект ангиотензина II, поддерживая нормальную величину артериального давления.
Синдром псевдо-Барттера может быть вызван продолжительным приемом диуретиков, длительной хлордефицитной диетой, периодически возникающей рвотой, чрезмерным приемом слабительных, муковисцидозом.
Симптомы
Синдром Барттера проявляется сразу после рождения или в раннем детском возрасте. Его клиническая картина обусловлена имеющимся хроническим дефицитом калия. Наблюдается полиурия и, как следствие, эксикоз (обезвоживание), поражение мышечной системы (слабость скелетных мышц, сердечной мышцы, гладкой мускулатуры, вялый псевдопаралич, судороги), отставание ребенка в умственном и физическом развитии, поражение нервной системы (парестезии и ригидность конечностей) при отсутствии артериальной гипертензии (нормальном или сниженном АД).
Неонатальный вариант патологии манифестирует в период внутриутробного развития плода многоводием, часто сопровождается преждевременными родами и имеет тяжелое течение. У недоношенных новорожденных наблюдается плохой аппетит, сонливость, быстрая потеря веса, задержка психомоторного развития, мышечная гипотония, нарушения зрения и слуха, гипертермия.
Классический тип синдрома проявляется в раннем детском возрасте (после 1 года жизни) задержкой роста и развития ребенка, полиурией, склонностью к дегидратации, рвотой, запорами, полидипсией. Синдром Гительмана выявляется примерно с 6-летнего возраста или позднее; характеризуется мышечной слабостью, утомляемостью, случаями возвратной тетании и имеет более доброкачественное течение.
При синдроме псевдо-Барттера развиваются аналогичные симптомы, обусловленные гипокалиемическим метаболическим алкалозом; данная патология часто встречается у молодых девушек, использующих для похудания диуретики и строго ограниченную диету.
Диагностика
Неонатальный тип синдрома на первой неделе жизни можно определить по наличию метаболического алкалоза с гипокалиемией, низкому удельному весу мочи, содержащей большое количество ионов K, Na, Cl, Ca, высокому уровню простагландинов в крови и моче, большой активности ренина и альдостерона в крови.
При классическом варианте течения выявляют гипокалиемический метаболический алкалоз с повышенным или нормальным содержанием кальция, не нарушенную способность концентрировать мочу. В случае синдрома Гительмана обнаруживается резко выраженная гипомагниемия и гипокальциурия. По этим показателям синдром Барттера диагностируется при исключении приема диуретиков и слабительных средств, потерь калия и хлоридов через ЖКТ.
В редких случаях возможно выполнение биопсии почки, которая позволяет выявить гиперплазию околоклубочкового аппарата. Патологию следует дифференцировать от хронической рвоты, злоупотребления мочегонными препаратами, состояний, связанных с дефицитом магния, изолированного гиперальдостеронизма, хронической надпочечниковой недостаточности.
Лечение синдрома Барттера
Традиционное лечение различных типов синдрома включает заместительную и медикаментозную терапию. Необходимо обеспечение достаточного поступления калия и хлорида натрия с пищей, дополнительный прием препаратов калия. В лечении неонатального вида патологии сразу же после рождения ребенка начинают экстренную интенсивную заместительную терапию с помощью инфузий солевых растворов (NaCl, KCl). Для уменьшения потери калия организмом назначают калийсберегающие диуретики (спиронолактон, триамтерен, амилорид).
Необходим прием ингибиторов синтеза простагландинов (НПВС: индометацина, аспирина) и ингибиторов АПФ (каптоприла), снижающих секрецию ренина и альдостерона. У недоношенных младенцев из-за побочного действия индометацина его применение необходимо отстрочить до достижения детьми 4-6 недельного возраста. Коррекцию гипомагниемии при синдроме Гительмана проводят препаратами магния. Для лечения синдрома псевдо-Барттера необходимо устранить первопричину заболевания.
Прогноз и профилактика
Ранняя диагностика и адекватное лечение классического синдрома Барттера позволяет уменьшить тяжесть проявлений, отставание в умственном и физическом развитии. При неонатальном типе заболевания в отсутствии своевременного лечения возможна гибель ребенка из-за тяжелых электролитных нарушений и дегидратации организма. При тяжелом и долгом клиническом течении заболевания часто развивается нефрокальциноз, который может привести к хронической почечной недостаточности. Профилактика не разработана.
Синдром Барттера — это заболевание почек
Синдром Барттера (тубулопатия) — это заболевание почек, которое относится к врожденным дефектам. Заболевание наследуется от обоих родителей. Это нарушение кислотно-щелочного баланса в организме в результате плохой работы почечных спиралей. Нарушения в фильтрации крови приводят к недостатку калия и натрия в организме.
Синдром Барттера — это группа генетически обусловленных заболеваний, называемых тубулопатиями. Это заболевания, связанные с нарушением резорбтивной или секреторной функции почечных канальцев, возникающие при нормальной или только минимально сниженной клубочковой фильтрации. Синдром Барттера чаще всего встречается в возрасте от одного до двух лет. Это редкое заболевание, и оно наследуется рецессивно, то есть ребенок должен получить по одной копии дефектного гена от каждого родителя. Мутации связаны с четырьмя генами, связанными с кодированием белка, ответственными за реабсорбцию.
При синдроме Барттера встречаются следующие симптомы:
Характеристика заболевания
Заболевание представляет собой нарушение реабсорбции натрия, калия и хлорида в клубочек, в Петля Генле, которая влечет за собой изменения в составе крови и проблемы с выработкой мочи. Клубочки — это сеть правильно построенных капилляров, через которые фильтруется кровь, вода и питательные вещества, что приводит к первичной мочи. Любое нарушение работы почек на этой стадии приводит к слишком большой потере ионов калии, натрии и хлора. Когда они выводятся из организма в чрезмерных количествах, существующий кислотно-щелочной баланс перестает существовать. Речь идет о метаболический алкалоз, то есть состояние повышенного pH в плазме крови, вызванное потерей ионов калия.
Что происходит потом? Когда клубочки истощают слишком много ионов калия и натрия, и в организме начинает не хватать циркулирующей крови, организм реагирует, увеличивая выработку гормонов — ренина, ангиотензина и альдостерона. Обычно этих соединений мало в организме, и они ответственны за снижение потери воды и кровяного давления. Но сейчас их избыток приводит к развитию болезни.
Синдром Барттера встречается в нескольких вариантах, в зависимости от того, какая комбинация генов повреждена и, следовательно, какой ионный канал не работает должным образом. Отдельные виды заболеваний, в том числе синдром Гительмана немного отличается по ходу, но общим для них является снижение уровня калия в крови (так называемая гипокалиемия). При одном из вариантов заболевания слух также может быть поврежден.
Симптомы
Диагностика и лечение
Врожденный синдром Барттера диагностируется у очень маленьких детей с помощью диагностических тестов. Вы также можете видеть, что ребенок набирает вес слишком медленно и плохо растет. Генетическое тестирование также может быть сделано, чтобы точно определить, какие гены были повреждены.
Как генетический дефект, синдром Барттера неизлечим. Однако ведется консервативное лечение. Он заключается в пероральном введении препаратов калия и пополнении уровня других электролитов в организме, в некоторых случаях даже внутривенно. В менее тяжелых состояниях это достаточно, чтобы больной вел достаточно нормальную жизнь. Более тяжелые случаи заболевания, когда возникает почечная недостаточность, требуют диализа и даже трансплантации почки.
Синдром Барттера и синдром Гительмана
(Синдром Барттера; синдром Гительмана)
, MD, Perelman School of Medicine at The University of Pennsylvania
Патофизиология
Синдром Барттера и более распространенный синдром Гительмана являются результатом
Нарушенная реабсорбция хлорида натрия
При синдроме Барттера дефект затрагивает восходящее толстое колено петли Генле. При синдроме Гительмана выявляется дефект дистальных канальцев.
При обоих синдромах ухудшение реабсорбции хлорида натрия вызывает легкую форму дегидратации, что приводит к увеличению высвобождения ренина и альдостерона, в результате чего происходит потеря калия и водорода. При синдроме Барттера возникает повышение секреции простагландина, а также нарушение концентрационной способности почек вследствие ухудшения образования медуллярного градиента концентрации. При синдроме Гительмана распространенными являются гипомагниемия и низкая экскреция кальция с мочой. При обоих расстройствах потеря натрия способствует легкому хроническому уменьшению объема плазмы, что проявляется нормальным или низким уровнем артериального давления, несмотря на высокие уровни ренина и ангиотензина.
Этиология
Справочные материалы по этиологии
1. Fulchiero R, Seo-Mayer P: Bartter syndrome and Gitelman syndrome. Pediatr Clin North Am 66(1):121–134, 2019. doi: 10.1016/j.pcl.2018.08.010
Клинические проявления
Синдром Барттера чаще манифестирует пренатально или же в младенчестве или раннем детстве. Синдром Гительмана проявляется в позднем детстве или в зрелом возрасте. Пренатально синдром Барттера может проявляться задержкой внутриутробного развития и многоводием. Различные формы синдрома Барттера могут иметь конкретные проявления, включая потерю слуха, гипокальциемию и нефрокальциноз, в зависимости от основного генетического дефекта. Дети с синдромом Барттера чаще, чем дети с синдромом Гительмана, могут рождаться преждевременно, иметь низкий рост и неудовлетворительное постнатальное развитие, также у некоторых детей имеется умственная отсталость.
У большинства пациентов артериальное давление низкое или на нижних пределах нормального, и они могут иметь признаки гиповолемии. Неспособность удержать в организме калий, кальций или магний может привести к мышечной слабости, спазмам, судорогам, тетании или усталости, особенно при синдроме Гительмана. Могут наблюдаться полидипсия, полиурия и рвота.
В общем, ни синдром Барттера, ни синдром Гительмана обычно не приводят к развитию хронической почечной недостаточности.
Диагностика
Уровни электролитов в сыворотке крови и моче



