Синдром гительмана что это
Синдром Гительмана (часто называемый вариантом синдрома Бартера) также представляет собой редкое аутосомно-рецессивное заболевание с гипокалиемическим метаболическим алкалозом. Отличительная особенность синдрома — гипокальциурия и гипомагниемия, в типичных случаях он проявляется в позднем детском или раннем зрелом возрасте.
Патогенез синдрома Гительмана. Биохимические сдвиги при синдроме Гительмана напоминают таковые при постоянном применении тиазидных диуретиков. Эти средства действуют на натрий-хлоридные котранспортеры, NCCT, локализованные в дистальных извитых канальцах. Генетические исследования обнаруживают у больных дефекты гена, кодирующего NCCT.
Клинические проявления синдрома Гительмана. Синдром Гительмана, как правило, проявляется в более позднем возрасте, чем синдром Бартера. В анамнезе больных часто имеются мышечные судороги и спазмы обусловленные, вероятно, низким уровнем магния в сыворотке. Повторные эпизоды обезвоживания обычно отсутствуют. Биохимические сдвиги включают гипокалиемию, метаболический ацидоз и гипомагниемию. Уровень кальция в моче резко снижен (в отличие от синдрома Бартера), а содержание магния повышено.
Уровень ренина и альдостерона, как правило, в норме; не увеличивается секреция простагландина Е. Отставание в росте выражено слабее, чем при синдроме Бартера. Диагностика. Синдром Гительмана диагностируют у подростков или взрослых при наличии гипокалиемического метаболического алкалоза, гипомагниемии и гипокальциурии. Лечение. Для коррекции гипокалиемии и гипомагниемии используют калиевые и магниевые добавки. Добавки натрия или ингибиторы синтеза простагландинов обычно не требуются, так как ни гиповолемия, ни повышение экскреции простагландина Е у больных, как правило, не наблюдается.
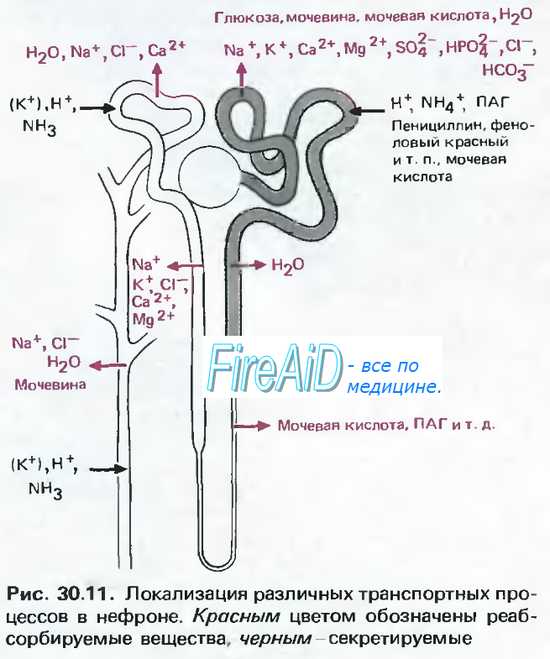
В настоящее время охарактеризованы врожденные дефекты различных транспортеров, функционирующих в разных отделах нефрона. Почечный канальцевый ацидоз и нефрогенный несахарный диабет подробно рассматривались соответственно. Аутосомно-рецессивная цистинурия наблюдается главным образом у выходцев со среднего Востока. Она характеризуется повторным образованием мочевых камней и обусловлена дефектом высокоаффинного транспортера L-цистина и двухосновных аминокислот в проксимальных почечных канальцах.
Для Х-сцепленной мочекаменной болезни (болезни Дента) также характерно повторное образование камней и прогрессирование до синдрома Фанкони. В соответствии со своим Х-сцепленным обследованием заболевание наблюдается почти включительно у мальчиков. В его основе лежит мутация гена, кодирующего потенциалзависимые каналы, CLN5, присутствующие во всех отделах нефрона. Активирующие или инактивирующие мутации гена кальциевых рецепторов (через которые ПТГ усиливает реабсорбцию кальция в петле Генле) обусловливают соответственно тяжелый гипо- или гиперпаратиреоз.
Активирующие мутации гена эпителиальных натриевых каналов, функционирующих в собирательных трубочках, лежат в основе наследственной формы артериальной гипертонии, синдрома Лидд-ла. У больных с этим синдромом конститутивно активирована реабсорбция натрия в собирательных трубочках, снижен уровень калия в сыворотке и подавлена секреция альдостерона. Инактивирующие мутации этого гена лежат в основе псевдогипоальдостеронизма, проявляющегося резкой потерей натрия и гиперкалиемией. Один из вариантов этого состояния характеризуется системными нарушениями, включая выделение хлорида с потом, и может напоминать муковисцидоз.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
![]()
СОДЕРЖАНИЕ
Признаки и симптомы
Фенотипические вариации, наблюдаемые у пациентов, вероятно, являются результатом различий в их генетическом фоне и могут зависеть от того, какая конкретная аминокислота в белке NCCT подверглась мутации. Исследование Riviera-Munoz et al. идентифицировали подмножество людей с синдромом Гительмана с тяжелым фенотипическим выражением. Клиническими проявлениями, наблюдаемыми в этой группе, были нервно-мышечные проявления, задержка роста и желудочковые аритмии. Пациенты были в основном мужчинами, и было обнаружено, что по крайней мере один аллель дефекта сплайсинга в гене SLC12A3.
Причина

Вторичным эффектом инактивированного котранспортера хлорида натрия является последующая активация ренин-ангиотензин-альдостероновой системы (РААС). Активация РААС является побочным продуктом неспособности дистальных извитых канальцев повторно поглощать электролиты, в частности натрия и хлорида, что приводит к клеточной дегидратации. RAAS пытается компенсировать это обезвоживание, приводящее к низкому уровню калия в сыворотке крови.
Небольшой процент случаев синдрома Гительмана можно объяснить мутациями в гене CLCNKB. Этот ген связан с функцией почечного хлоридного канала CLC-Kb, расположенного на базолатеральной мембране клеток в толстой восходящей конечности петли Генле. Генетические вариации или мутации в CLCNKB изначально были связаны с классическим синдромом Барттера. Если в гене SLC12A3 не обнаружены мутации, можно провести скрининг, чтобы исключить участие гена CLCNKB.
Синдром Гительмана наследуется по аутосомно- рецессивному типу: по одному дефектному аллелю должен быть унаследован от каждого родителя.
В 2021 году было описано, что мутации тРНК, кодирующих изолейцин (MT-TI) и фенилаланин (MT-TF) в митохондриальной ДНК, вызывают синдром Гительмана. Эти гомомплазматические мутации мтДНК наследуются по материнской линии.
Диагностика
Ключевым моментом является обследование для исключения дифференциальной диагностики электролитных нарушений.
У пациентов с ранним началом заболевания, таких как младенцы и дети, индометацин является препаратом выбора, используемым для лечения нарушений роста. Индометацин в исследовании Blanchard et. al 2015 показал повышение уровня калия в сыворотке и снижение концентрации ренина. Побочные эффекты индометацина включают снижение скорости клубочковой фильтрации и желудочно-кишечные расстройства.
Эпидемиология
Синдром Гительмана встречается у 1 из 40 000 гомозиготных людей. Соотношение мужчин и женщин составляет 1: 1. Это заболевание обычно возникает после первого десятилетия жизни, в подростковом или взрослом возрасте, но может возникать и в неонатальном периоде. Гетерозиготные носители мутации гена SLC12A3 составляют 1% населения. Родители с синдромом Гительмана имеют низкую вероятность передачи заболевания своему потомству, примерно 1 из 400, если только они оба не являются носителями болезни.
История
Синдром Барттера и синдром Гительмана
(Синдром Барттера; синдром Гительмана)
, MD, Perelman School of Medicine at The University of Pennsylvania
Патофизиология
Синдром Барттера и более распространенный синдром Гительмана являются результатом
Нарушенная реабсорбция хлорида натрия
При синдроме Барттера дефект затрагивает восходящее толстое колено петли Генле. При синдроме Гительмана выявляется дефект дистальных канальцев.
При обоих синдромах ухудшение реабсорбции хлорида натрия вызывает легкую форму дегидратации, что приводит к увеличению высвобождения ренина и альдостерона, в результате чего происходит потеря калия и водорода. При синдроме Барттера возникает повышение секреции простагландина, а также нарушение концентрационной способности почек вследствие ухудшения образования медуллярного градиента концентрации. При синдроме Гительмана распространенными являются гипомагниемия и низкая экскреция кальция с мочой. При обоих расстройствах потеря натрия способствует легкому хроническому уменьшению объема плазмы, что проявляется нормальным или низким уровнем артериального давления, несмотря на высокие уровни ренина и ангиотензина.
Этиология
Справочные материалы по этиологии
1. Fulchiero R, Seo-Mayer P: Bartter syndrome and Gitelman syndrome. Pediatr Clin North Am 66(1):121–134, 2019. doi: 10.1016/j.pcl.2018.08.010
Клинические проявления
Синдром Барттера чаще манифестирует пренатально или же в младенчестве или раннем детстве. Синдром Гительмана проявляется в позднем детстве или в зрелом возрасте. Пренатально синдром Барттера может проявляться задержкой внутриутробного развития и многоводием. Различные формы синдрома Барттера могут иметь конкретные проявления, включая потерю слуха, гипокальциемию и нефрокальциноз, в зависимости от основного генетического дефекта. Дети с синдромом Барттера чаще, чем дети с синдромом Гительмана, могут рождаться преждевременно, иметь низкий рост и неудовлетворительное постнатальное развитие, также у некоторых детей имеется умственная отсталость.
У большинства пациентов артериальное давление низкое или на нижних пределах нормального, и они могут иметь признаки гиповолемии. Неспособность удержать в организме калий, кальций или магний может привести к мышечной слабости, спазмам, судорогам, тетании или усталости, особенно при синдроме Гительмана. Могут наблюдаться полидипсия, полиурия и рвота.
В общем, ни синдром Барттера, ни синдром Гительмана обычно не приводят к развитию хронической почечной недостаточности.
Диагностика
Уровни электролитов в сыворотке крови и моче
Синдром Барттера

Синдром Барттера – это генетически обусловленная тубулопатия, проявляющаяся выраженными нарушениями электролитного обмена (гипокалиемией), кислотно-щелочного равновесия (метаболическим алкалозом), гиповолемией, компенсаторной гиперплазией юкстагломерулярного (околоклубочкового) аппарата почек и вторичным гиперальдостеронизмом. Диагностируется по клинической симптоматике: полиурии, отставании в психомоторном развитии, гипотонии мышц, а также лабораторным показателям крови и мочи. Лечение заключается в заместительной терапии препаратами калия, натрия и магния, приеме калийсберегающих диуретиков, ингибиторов синтеза простагландинов и АПФ.

Общие сведения
Причины
Причиной синдрома Барттера считают нарушение транспортной функции почечных канальцев, проявляющееся снижением реабсорбции ионов Cl (и, соответственно, Na) клетками восходящего отдела петли Генле. Это приводит к гиповолемии, избытку натрия и воды в дистальной части нефрона, усилению секреции ионов K и натрий-калиевого обмена. Гипокалиемия стимулирует, в свою очередь, образование простагландинов Е2 и I2, приводящее к усилению секреции ренина и ангиотензина II.
Хроническая гиперренинемия способствует развитию гиперплазии юкстагломерулярного аппарата почек и повышенной продукции альдостерона надпочечниками. Ангиотензин II и альдостерон вызывают увеличение уровня почечного калликреина с дальнейшим повышением содержания брадикинина плазмы крови. Альдостерон приводит к усилению выведения калия почками. Калликреин (брадикинин) и простагландины блокируют вазопрессорный эффект ангиотензина II, поддерживая нормальную величину артериального давления.
Синдром псевдо-Барттера может быть вызван продолжительным приемом диуретиков, длительной хлордефицитной диетой, периодически возникающей рвотой, чрезмерным приемом слабительных, муковисцидозом.
Симптомы
Синдром Барттера проявляется сразу после рождения или в раннем детском возрасте. Его клиническая картина обусловлена имеющимся хроническим дефицитом калия. Наблюдается полиурия и, как следствие, эксикоз (обезвоживание), поражение мышечной системы (слабость скелетных мышц, сердечной мышцы, гладкой мускулатуры, вялый псевдопаралич, судороги), отставание ребенка в умственном и физическом развитии, поражение нервной системы (парестезии и ригидность конечностей) при отсутствии артериальной гипертензии (нормальном или сниженном АД).
Неонатальный вариант патологии манифестирует в период внутриутробного развития плода многоводием, часто сопровождается преждевременными родами и имеет тяжелое течение. У недоношенных новорожденных наблюдается плохой аппетит, сонливость, быстрая потеря веса, задержка психомоторного развития, мышечная гипотония, нарушения зрения и слуха, гипертермия.
Классический тип синдрома проявляется в раннем детском возрасте (после 1 года жизни) задержкой роста и развития ребенка, полиурией, склонностью к дегидратации, рвотой, запорами, полидипсией. Синдром Гительмана выявляется примерно с 6-летнего возраста или позднее; характеризуется мышечной слабостью, утомляемостью, случаями возвратной тетании и имеет более доброкачественное течение.
При синдроме псевдо-Барттера развиваются аналогичные симптомы, обусловленные гипокалиемическим метаболическим алкалозом; данная патология часто встречается у молодых девушек, использующих для похудания диуретики и строго ограниченную диету.
Диагностика
Неонатальный тип синдрома на первой неделе жизни можно определить по наличию метаболического алкалоза с гипокалиемией, низкому удельному весу мочи, содержащей большое количество ионов K, Na, Cl, Ca, высокому уровню простагландинов в крови и моче, большой активности ренина и альдостерона в крови.
При классическом варианте течения выявляют гипокалиемический метаболический алкалоз с повышенным или нормальным содержанием кальция, не нарушенную способность концентрировать мочу. В случае синдрома Гительмана обнаруживается резко выраженная гипомагниемия и гипокальциурия. По этим показателям синдром Барттера диагностируется при исключении приема диуретиков и слабительных средств, потерь калия и хлоридов через ЖКТ.
В редких случаях возможно выполнение биопсии почки, которая позволяет выявить гиперплазию околоклубочкового аппарата. Патологию следует дифференцировать от хронической рвоты, злоупотребления мочегонными препаратами, состояний, связанных с дефицитом магния, изолированного гиперальдостеронизма, хронической надпочечниковой недостаточности.
Лечение синдрома Барттера
Традиционное лечение различных типов синдрома включает заместительную и медикаментозную терапию. Необходимо обеспечение достаточного поступления калия и хлорида натрия с пищей, дополнительный прием препаратов калия. В лечении неонатального вида патологии сразу же после рождения ребенка начинают экстренную интенсивную заместительную терапию с помощью инфузий солевых растворов (NaCl, KCl). Для уменьшения потери калия организмом назначают калийсберегающие диуретики (спиронолактон, триамтерен, амилорид).
Необходим прием ингибиторов синтеза простагландинов (НПВС: индометацина, аспирина) и ингибиторов АПФ (каптоприла), снижающих секрецию ренина и альдостерона. У недоношенных младенцев из-за побочного действия индометацина его применение необходимо отстрочить до достижения детьми 4-6 недельного возраста. Коррекцию гипомагниемии при синдроме Гительмана проводят препаратами магния. Для лечения синдрома псевдо-Барттера необходимо устранить первопричину заболевания.
Прогноз и профилактика
Ранняя диагностика и адекватное лечение классического синдрома Барттера позволяет уменьшить тяжесть проявлений, отставание в умственном и физическом развитии. При неонатальном типе заболевания в отсутствии своевременного лечения возможна гибель ребенка из-за тяжелых электролитных нарушений и дегидратации организма. При тяжелом и долгом клиническом течении заболевания часто развивается нефрокальциноз, который может привести к хронической почечной недостаточности. Профилактика не разработана.
![]()
СОДЕРЖАНИЕ
Признаки и симптомы
Фенотипические вариации, наблюдаемые у пациентов, вероятно, являются результатом различий в их генетическом фоне и могут зависеть от того, какая конкретная аминокислота в белке NCCT подверглась мутации. Исследование Riviera-Munoz et al. идентифицировали подмножество людей с синдромом Гительмана с тяжелым фенотипическим выражением. Клиническими проявлениями, наблюдаемыми в этой группе, были нервно-мышечные проявления, задержка роста и желудочковые аритмии. Пациенты были в основном мужчинами, и было обнаружено, что по крайней мере один аллель дефекта сплайсинга в гене SLC12A3.
Причина
Вторичным эффектом инактивированного котранспортера хлорида натрия является последующая активация ренин-ангиотензин-альдостероновой системы (РААС). Активация РААС является побочным продуктом неспособности дистальных извитых канальцев повторно поглощать электролиты, в частности натрия и хлорида, что приводит к клеточной дегидратации. RAAS пытается компенсировать это обезвоживание, приводящее к низкому уровню калия в сыворотке крови.
Небольшой процент случаев синдрома Гительмана можно объяснить мутациями в гене CLCNKB. Этот ген связан с функцией почечного хлоридного канала CLC-Kb, расположенного на базолатеральной мембране клеток в толстой восходящей конечности петли Генле. Генетические вариации или мутации в CLCNKB изначально были связаны с классическим синдромом Барттера. Если в гене SLC12A3 не обнаружены мутации, можно провести скрининг, чтобы исключить участие гена CLCNKB.
Синдром Гительмана наследуется по аутосомно- рецессивному типу: по одному дефектному аллелю должен быть унаследован от каждого родителя.
В 2021 году было описано, что мутации тРНК, кодирующих изолейцин (MT-TI) и фенилаланин (MT-TF) в митохондриальной ДНК, вызывают синдром Гительмана. Эти гомомплазматические мутации мтДНК наследуются по материнской линии.
Диагностика
Ключевым моментом является обследование для исключения дифференциальной диагностики электролитных нарушений.
У пациентов с ранним началом заболевания, таких как младенцы и дети, индометацин является препаратом выбора, используемым для лечения нарушений роста. Индометацин в исследовании Blanchard et. al 2015 показал повышение уровня калия в сыворотке и снижение концентрации ренина. Побочные эффекты индометацина включают снижение скорости клубочковой фильтрации и желудочно-кишечные расстройства.
Эпидемиология
Синдром Гительмана встречается у 1 из 40 000 гомозиготных людей. Соотношение мужчин и женщин составляет 1: 1. Это заболевание обычно возникает после первого десятилетия жизни, в подростковом или взрослом возрасте, но может возникать и в неонатальном периоде. Гетерозиготные носители мутации гена SLC12A3 составляют 1% населения. Родители с синдромом Гительмана имеют низкую вероятность передачи заболевания своему потомству, примерно 1 из 400, если только они оба не являются носителями болезни.



